6. Walkthrough of pre-constructed pipeline¶
6.1. List of pipelines¶
- WGS data analysis Enables the user to identify variants from raw sequencing reads and functionally annotate them. Multiple tools are integrated in the WGS analysis pipeline.
- RNA-Seq data analysis RNA-Seq part enables quantification of gene expression. The pipeline provides a final output of a list of differentially expressed genes. (to be constructed as provided in the CANEapp manual)
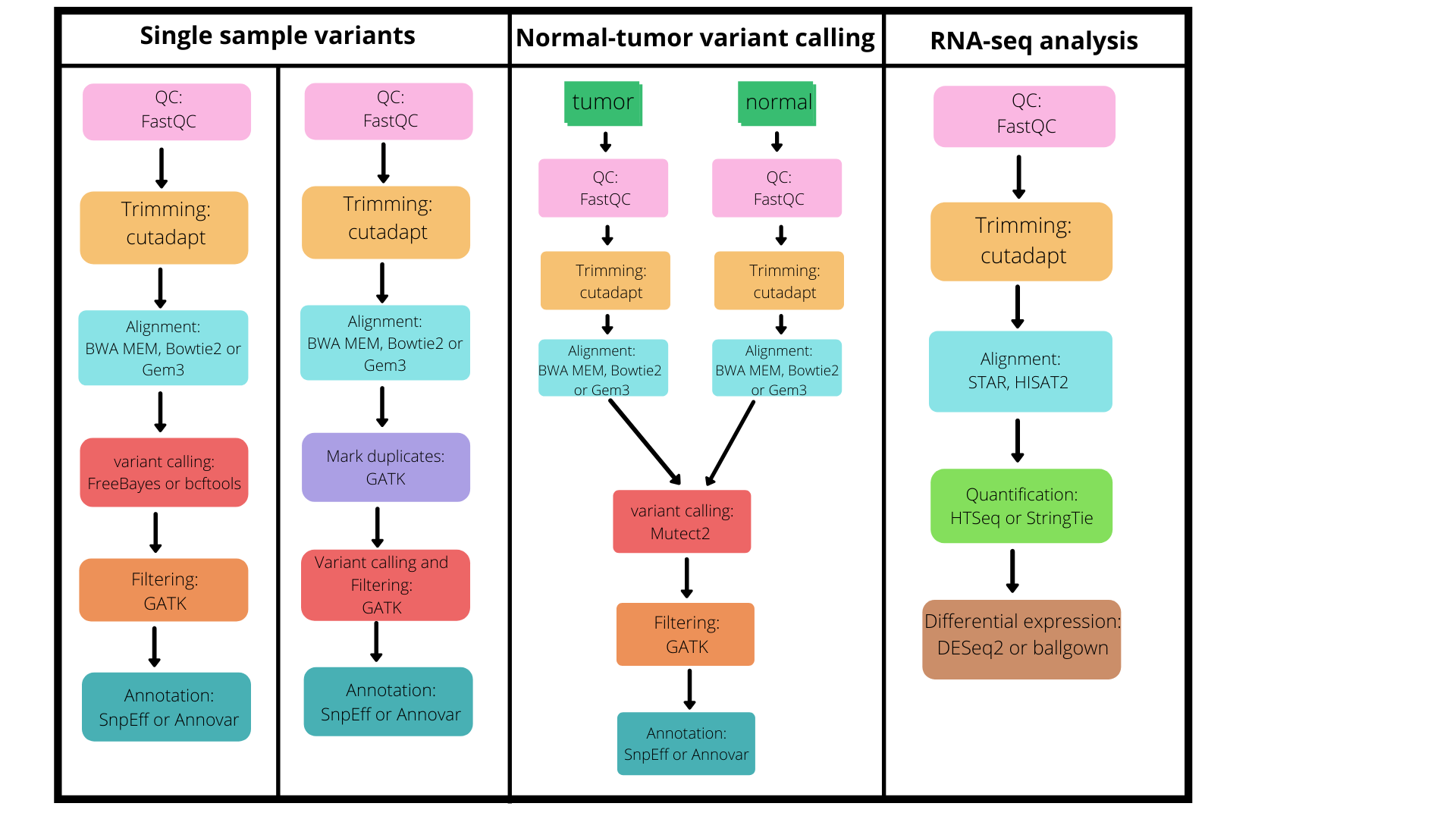 Figure 1: The figure below depicts the available pipelines supported by iCOMIC
Figure 1: The figure below depicts the available pipelines supported by iCOMIC
6.2. Description of the tools used¶
Table shows the tools incorporated in iCOMIC
| Function | DNA-Seq Tools | RNA-Seq Tools |
|---|---|---|
| Quality Control | FastQC, MultiQC, Cutadapt | FastQC, MultiQC, Cutadapt |
| Alignment | GEM-Mapper, BWA-MEM, Bowtie2 | STAR, HISAT2 |
| Variant Calling | GATK HC, samtools mpileup, FreeBayes, GATK Mutect2 | - |
| Annotation | Annovar, SnpEff | - |
| Expression Modeller | - | StringTie, HTSeq |
| Differential Expression | - | DESeq2, ballgown |
6.3. Tools for Quality Control¶
FastQC¶
It is a popular tool that can be used to provide an overview of the basic quality control metrics for raw next generation sequencing data. There are a number different analyses (called modules) that may be performed on a sequence data set. It provides summary graphs enabling the user to decide on the directions for further analysis.
MultiQC¶
MultiQC is a modular tool to aggregate results from bioinformatics analyses across multiple samples into a single report. It collects numerical stats from different modules enabling the user to track the behavior of the data in an efficient manner.
Cutadapt¶
Cutadapt is a trimming tool that enables the user to remove adapter and primer sequences in an error-tolerant manner. It can also aid in demultiplexing, filtering and modification of single-end and paired-end reads. Essential parameters for the tool are listed below. The detailed list of parameters of the tool are available in Cutadapt documentation.
| Parameter | Description |
|---|---|
-a |
3’ Adapter sequence |
-g |
5’ adapter sequence |
-Z |
Compression level |
-u (n) |
Removes n reads unconditionally |
-q |
Quality cutoff |
Aligners¶
GEM-Mapper¶
It is a high-performance mapping tool that performs alignment of sequencing reads against large reference genomes. GEM Mapper has been identified as an efficient mapping tool by a benchmarking analysis performed along with this study. Listed below are some parameters of the tool GEM-Mapper. Other parameters can be found in GEM-Mapper github page
| Parameter | Description |
|---|---|
-t |
Threads |
-e |
--alignment-max-error |
--alignment-global-min-identity |
Minimum global-alignment identity required |
--alignment-global-min-score |
Minimum global-alignment score required |
BWA-MEM¶
One of the most commonly used aligners available. It is identified as a faster and accurate algorithm among the algorithms in BWA software package. It is known for aligning long sequence query reads to the reference genome and also performs chimeric alignment. The parameters for BWA-MEM include the following. The other parameters for the tool can be found in BWA manual page
| Parameter | Description |
|---|---|
-t |
Threads |
-k |
minSeedLength |
-w |
Band width |
-d |
Z-dropoff |
-r |
seedSplitRatio |
-A |
matchScore |
Bowtie2¶
Bowtie2 is a fast and efficient algorithm for aligning reads to a reference sequence. It comprises various modes wherein it supports local, paired-end and gapped alignment. The key parameters for Bowtie2 include the following. All parameters for Bowtie2 are listed in Bowtie2 manual.
| Parameter | Description |
|---|---|
--threads |
Threads |
--cutoff (n) |
Index only the first (n) bases of the reference sequences (cumulative across sequences) and ignore the rest. |
-seed |
The seed for pseudo-random number generator |
-N |
Sets the number of mismatches to allowed in a seed alignment during multiseed alignment |
dvc |
the period for the difference-cover sample |
STAR¶
STAR is a rapid RNA-Seq read aligner specializing in fusion read and splice junction detection. Important parameters for STAR is given below. The other parameters for the tool can be found in STAR manual page
| parameters | Description |
|---|---|
-- runThreadN |
NumberOfThreads |
--runMode |
genomeGenerate |
--genomeDir |
/path/to/genomeDir |
--genomeFastaFiles |
/path/to/genome/fasta1 /path/to/genome/fasta2 |
--sjdbGTFfile |
/path/to/annotations.gtf |
--sjdbOverhang |
ReadLength-1 |
HISAT2¶
It is a fast and sensitive alignment program applicable for both RNA-seq and Whole-Genome Sequencing data and is known for rapid and accurate alignment of sequence reads to a single reference genome. The key parameters for the tool are given below. The other parameters for the tool can be found in HISAT2 manual page
| parameters | Description |
|---|---|
- x (hisat-idx) |
The basename of the index for the reference genome |
-q |
Reads which are FASTQ files |
--n-ceil (func) |
Sets a function governing the maximum number of ambiguous characters (usually Ns and/or .s) allowed in a read as a function of read length |
--ma (int) |
Sets the match bonus |
--pen-cansplice (int) |
Sets the penalty for each pair of canonical splice sites (e.g. GT/AG) |
Variant Callers¶
GATK HC¶
Variant caller for single sample analysis
One of the extensively used variant callers. Calls variants from the aligned reds corresponding to the reference genome. Some of the parameters for GATK Haplotype caller are listed beow. The complete parameter list is available at GATK Haplotypecaller article page
| Parameters | Description |
|---|---|
-contamination |
Contamination fraction to filter |
-hets |
heterozygosity |
-mbq |
Min base quality score |
-minReadsPerAlignStart |
Min Reads Per Alignment Start |
Samtools mpileup¶
Variant caller for single sample analysis
Samtools mpileup together with BCFtools call identifies the variants. Some key parameters to look are listed below. Parameters in detail are found in Samtools-mpileup manual page
| Parameter | Description |
|---|---|
-d |
--max-depth |
-q |
Minimum mapping quality for an alignment to be used |
-Q |
Minimum base quality for a base to be considered |
freebayes¶
Variant caller for single sample analysis
FreeBayes is a variant detector developed to identify SNPs, Indels, MNPs and complex variants with respect to the reference genome. Key parameters for FreeBayes are listed below. Other parameters can be found in detain in FreeBayes parameter page
| Parameter | Description |
|---|---|
-4 |
Include duplicate-marked alignments in the analysis. |
-m |
minimum mapping quality |
-q |
minimum base quality |
-! |
minimum coverage |
-U |
read mismatch limit |
GATK Mutect2¶
Variant caller for normal-tumor sample analysis
This tool identifies somatic mutations such as indels and SNAs in a diseased sample compared to the provided normal sample, using the haplotype assembly strategy. Parameters specific to Mutect2 include the following. The complete parameter list is available at GATK Mutect2 manual page
| Parameter | Description |
|---|---|
--base-quality-score-threshold |
Base qualities below this threshold will be reduced to the minimum |
--callable-depth |
Minimum depth to be considered callable for Mutect stats. Does not affect genotyping. |
--max-reads-per-alignment-start |
Maximum number of reads to retain per alignment start position. |
-mbq |
Minimum base quality required to consider a base for calling |
Annotators¶
SnpEff¶
SnpEff tool performs genomic variant annotations and functional effect prediction. Key parameters for the tool SnpEff are listed below. Detailed list of parameters is given in SnpEff manual page
| Parameter | Description |
|---|---|
-t |
Use multiple threads |
-cancer |
perform 'cancer' comparisons (Somatic vs Germline) |
-q |
Quiet mode |
-v |
Verbose mode |
-csvStats |
Create CSV summary file instead of HTML |
Annovar¶
Annovar can be used to efficiently annotate functional variants such as SNVs and indels, detected from diverse genomes. The tool also provides the user with multiple annotation strategies namely Gene-based, region-based and filter-based. Key parameters for Annovar include the following. Details of the tool can be found in Annovar documentation page
| Parameter | Description |
|---|---|
--splicing_threshold |
distance between splicing variants and exon/intron boundary |
--maf_threshold |
filter 1000G variants with MAF above this threshold |
--maxgenethread |
max number of threads for gene-based annotation |
--batchsize |
batch size for processing variants per batch (default: 5m) |
Expression modellers¶
StringTie¶
StringTie is known for efficient and rapid assembly of RNA-Seq alignments into possible transcripts. It employs a novel network flow algorithm and an optional de novo assembly algorithm to assemble the alignments. The important parameters to look into are listed below. The other parameters for the tool can be found in StringTie Manual Page
| Parameters | Description |
|---|---|
--rf |
Assumes a stranded library fr-firststrand |
--fr |
Assumes a stranded library fr-secondstrand |
--ptf (f_tab) |
Loads a list of point-features from a text feature file (f_tab) to guide the transcriptome assembly |
-l (label) |
Sets (label) as the prefix for the name of the output transcripts |
-m (int) |
Sets the minimum length allowed for the predicted transcripts |
HTSeq¶
HTSeq facilitates in counting the number of mapped reads to each gene. It provides the user with multiple modes of usage and also allows the creation of custom scripts. Key parameters for the tool are given below. The other parameters for the tool can be found in HTSeq Manual Page
| Parameters | Description |
|---|---|
-f |
Format of the input data |
-r |
For paired-end data, the alignment have to be sorted either by read name or by alignment position |
-s |
whether the data is from a strand-specific assay |
-a |
skip all reads with alignment quality lower than the given minimum value |
-m |
Mode to handle reads overlapping more than one feature |
Differential Expression tools¶
ballgown¶
ballgown is an R language based tool that enables the statistical analysis of assembled transcripts and differential expression analysis along with its visualization. Key arguments for the tool are given below. The other arguments for the tool can be found in ballgown Manual page
| Arguments | Description |
|---|---|
samples |
vector of file paths to folders containing sample-specific ballgown data |
dataDir |
file path to top-level directory containing sample-specific folders with ballgown data in them |
samplePattern |
regular expression identifying the subdirectories of\ dataDir containing data to be loaded into the ballgown object |
bamfiles |
optional vector of file paths to read alignment files for each sample |
pData |
optional data.frame with rows corresponding to samples and columns corresponding to phenotypic variables |
meas |
character vector containing either "all" or one or more of: "rcount", "ucount", "mrcount", "cov", "cov_sd", "mcov", "mcov_sd", or "FPKM" |
DESeq2¶
It Uses negative binomial distribution for testing differential expression using R language. Some of the arguments to look into are given below. The other arguments for the tool can be found in DESeq2 Manual Page
| Arguments | Description |
|---|---|
object |
A Ranged Summarized Experiment or DESeqDataSet |
groupby |
a grouping factor, as long as the columns of object |
run |
optional, the names of each unique column in object |
renameCols |
whether to rename the columns of the returned object using the levels of the grouping factor |
value |
an integer matrix |